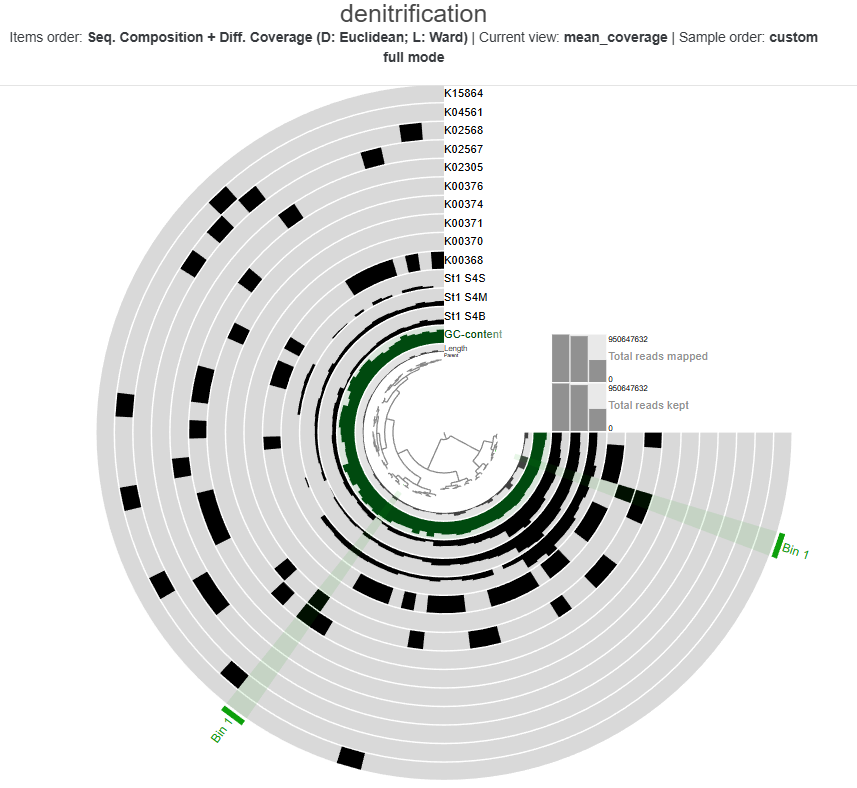
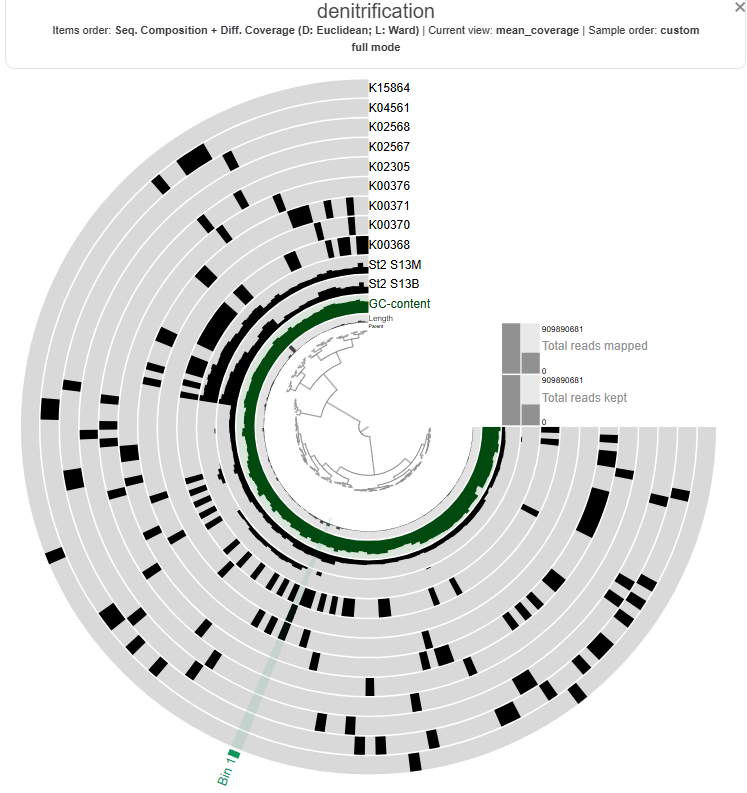
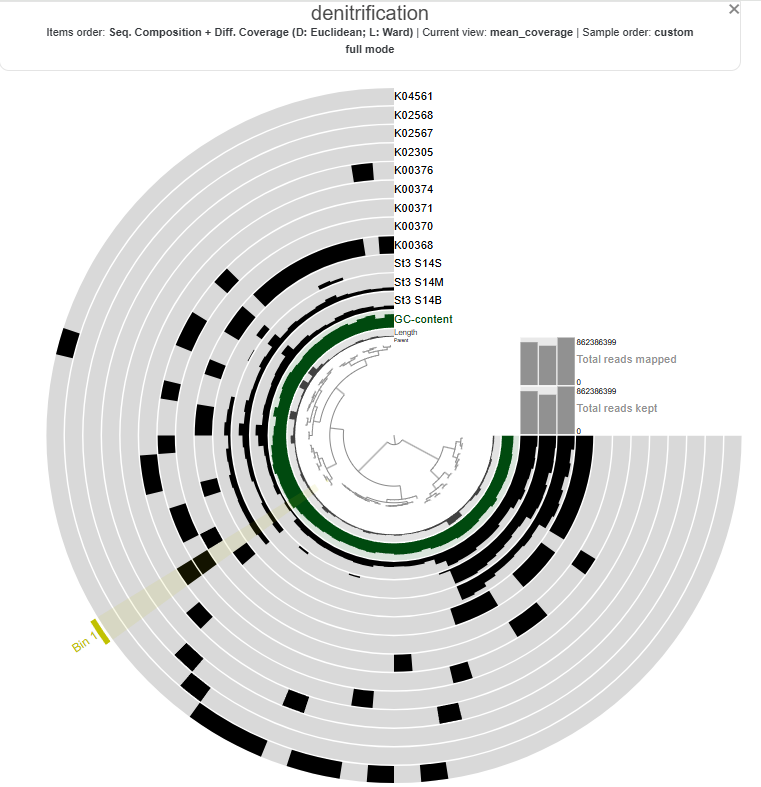
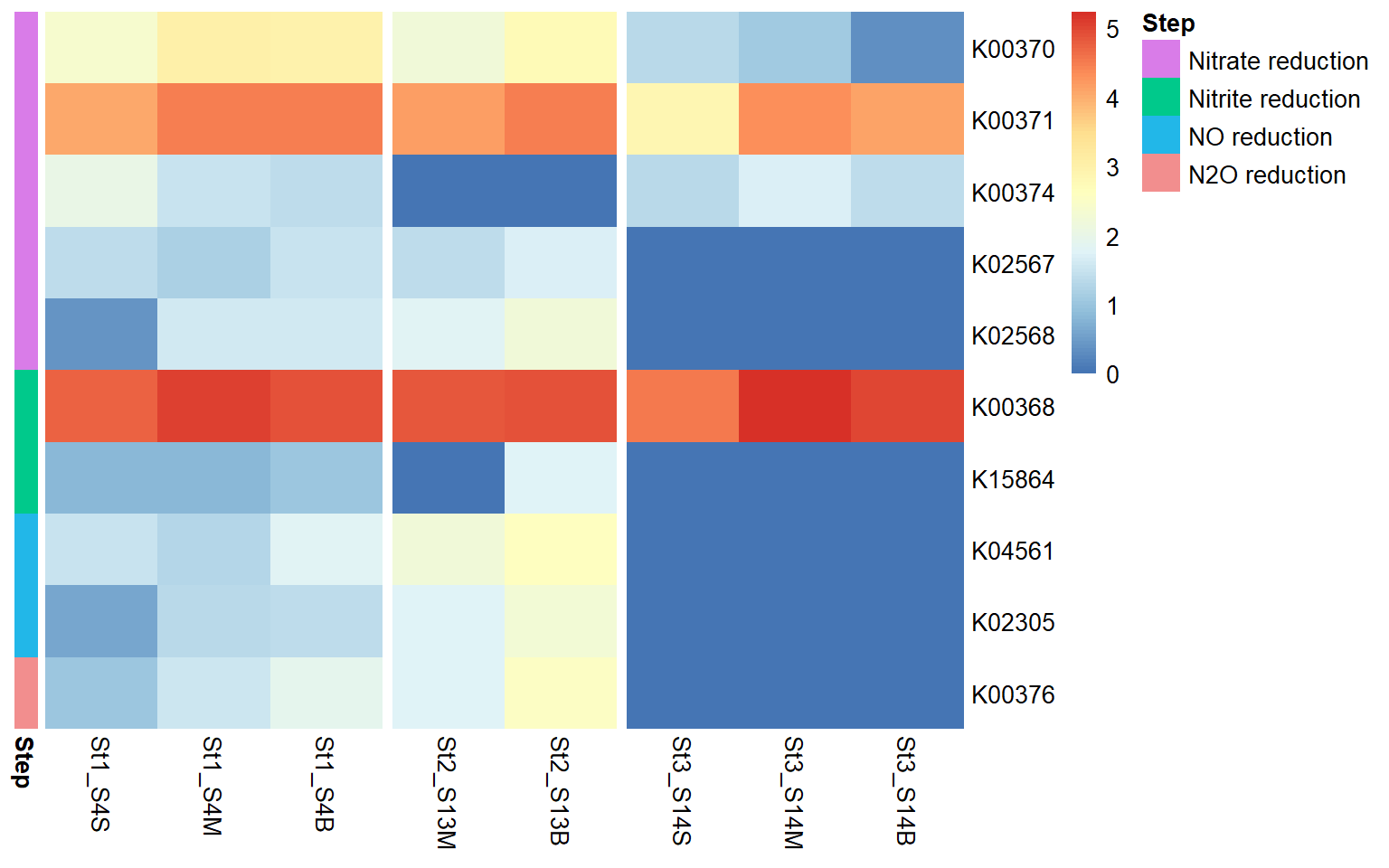
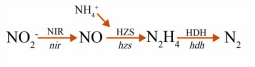
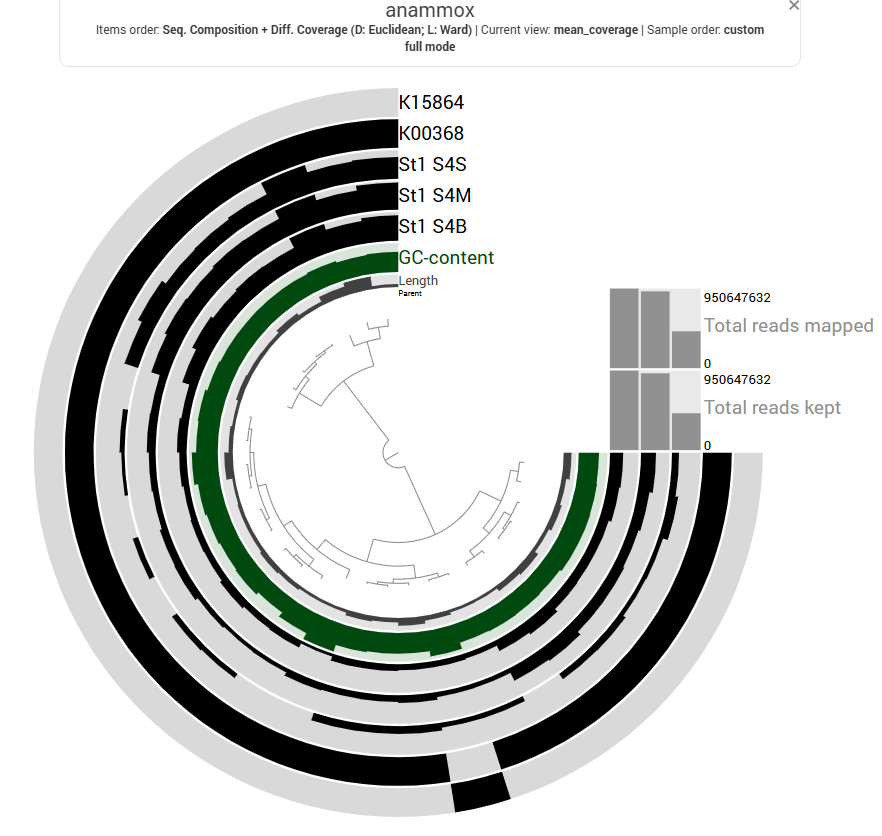
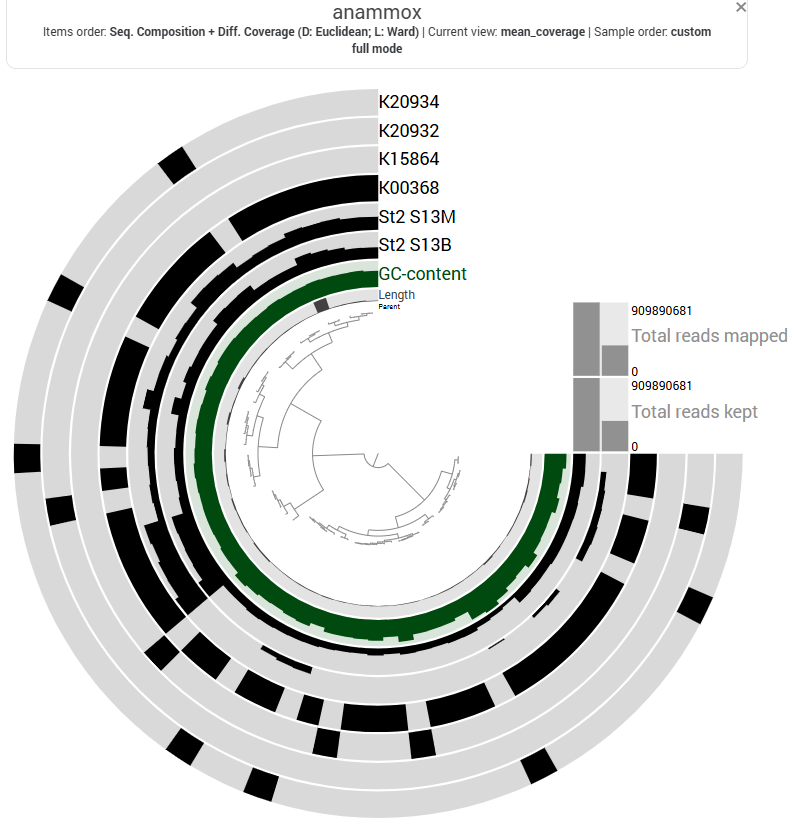
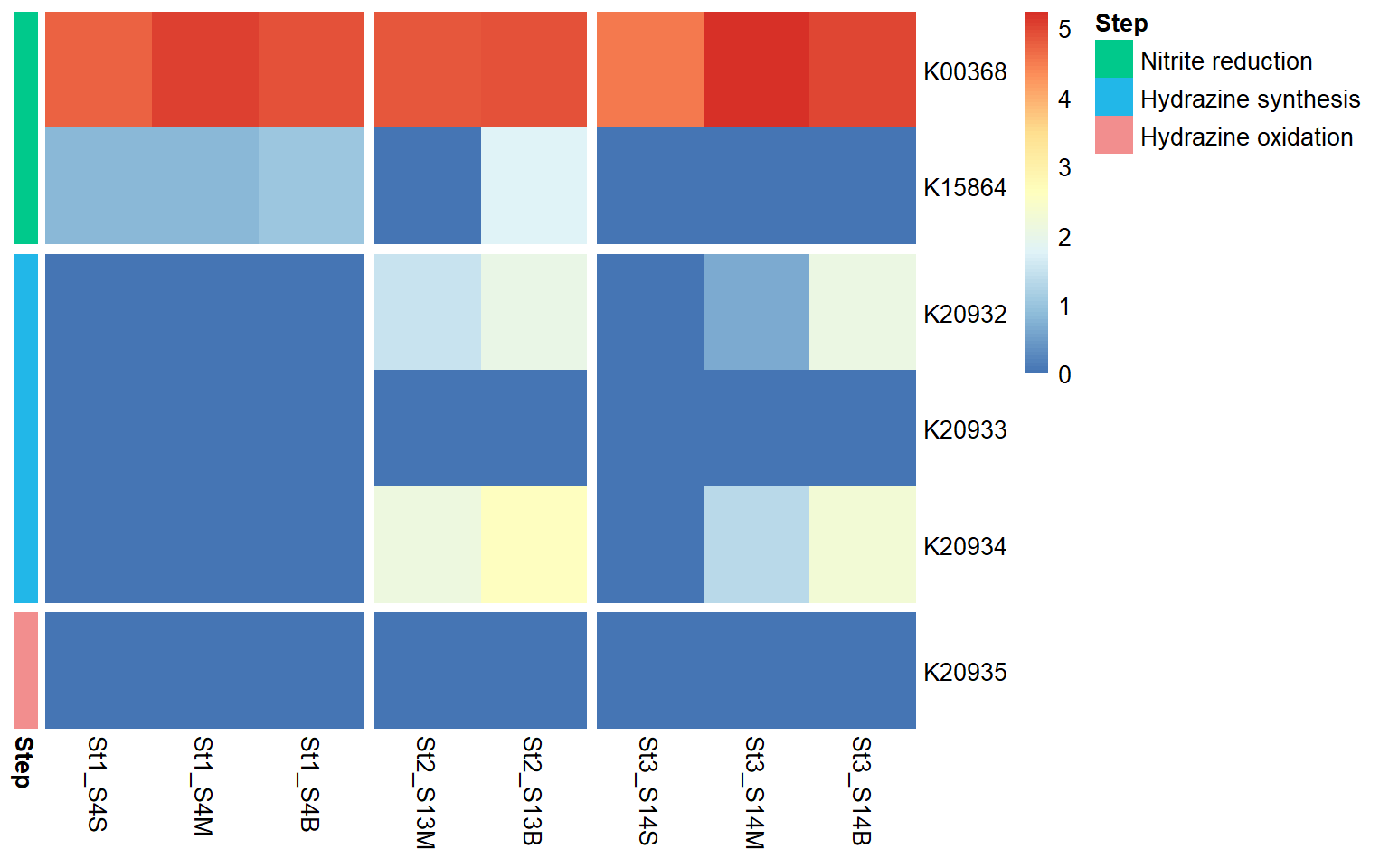
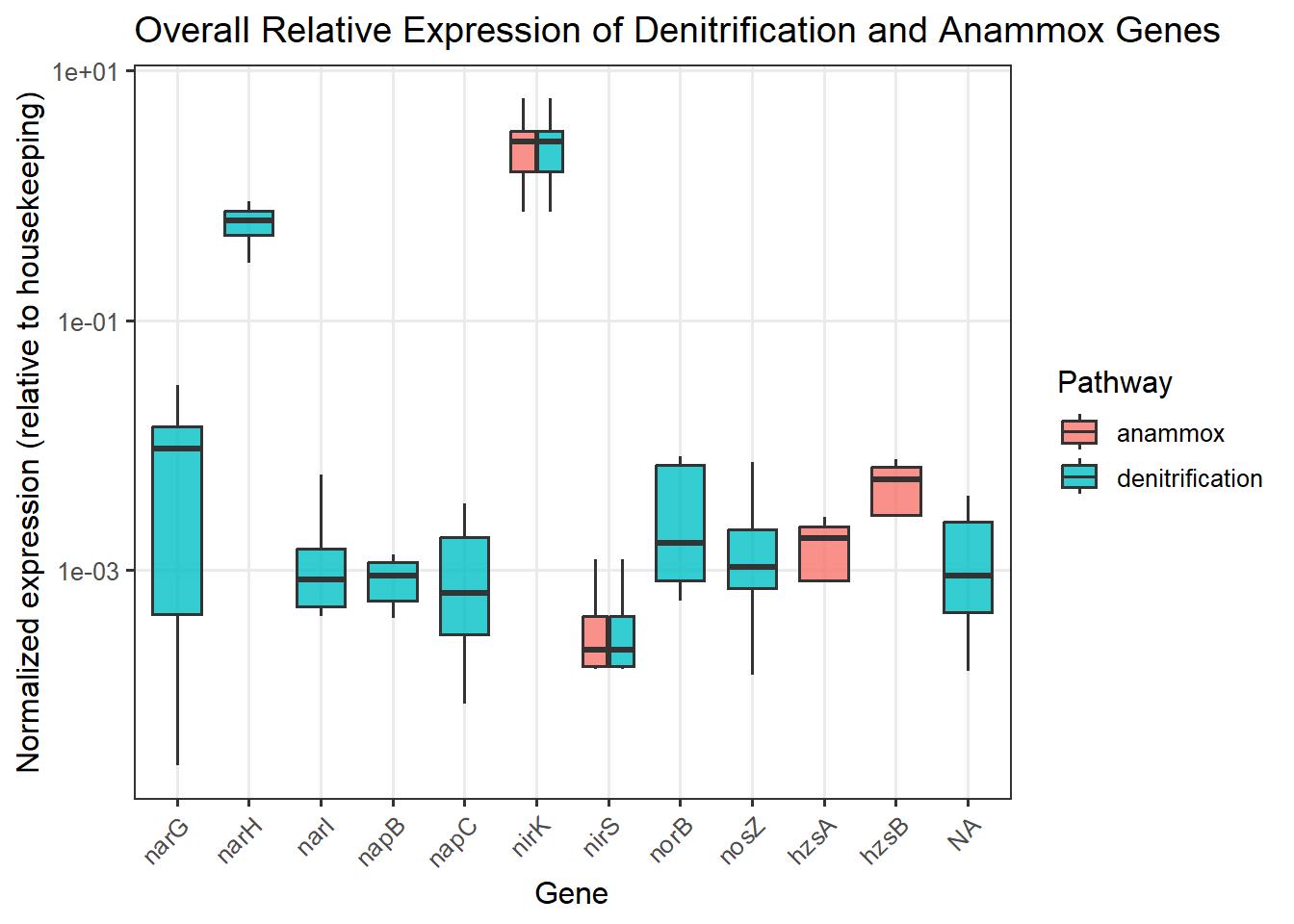
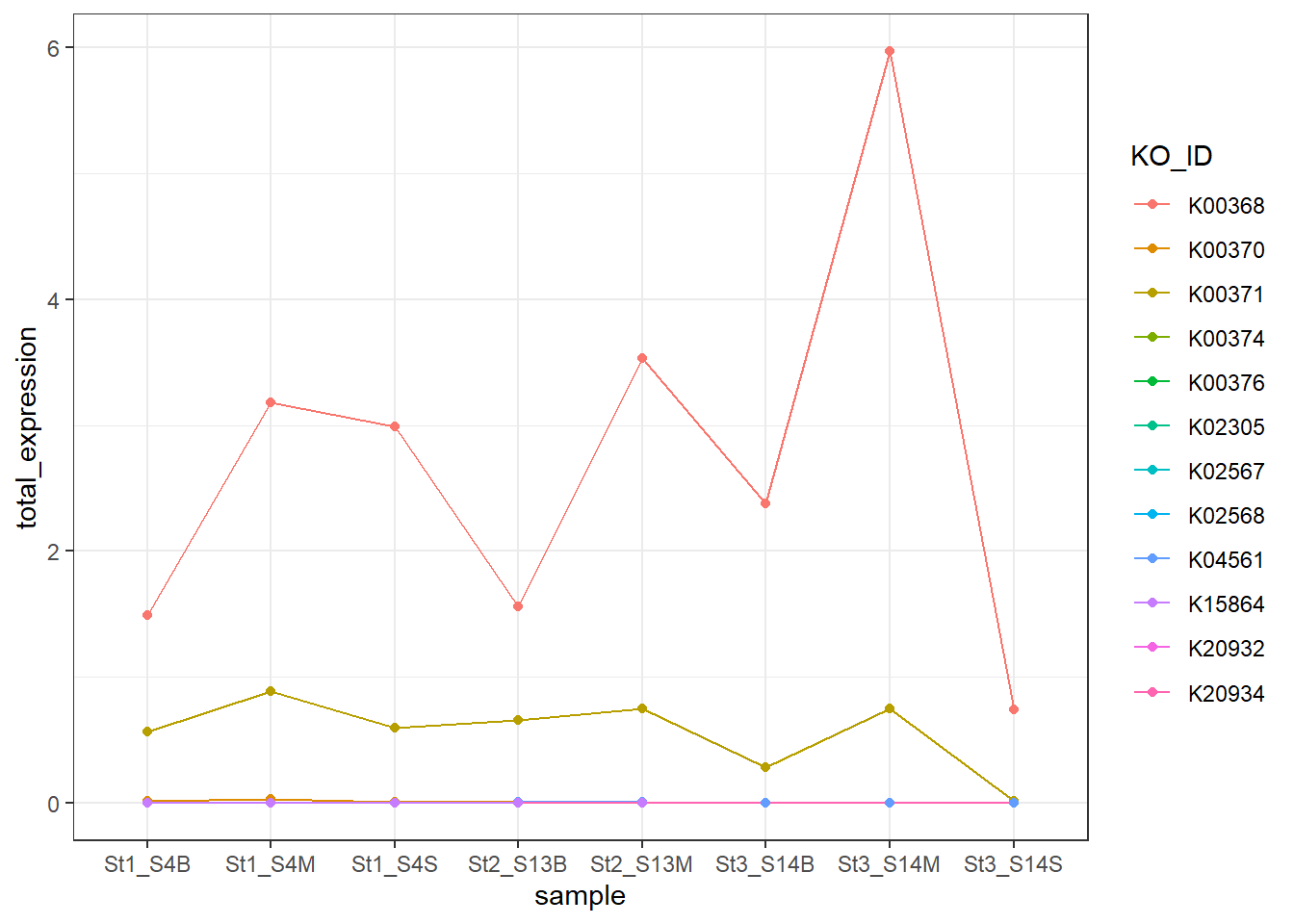
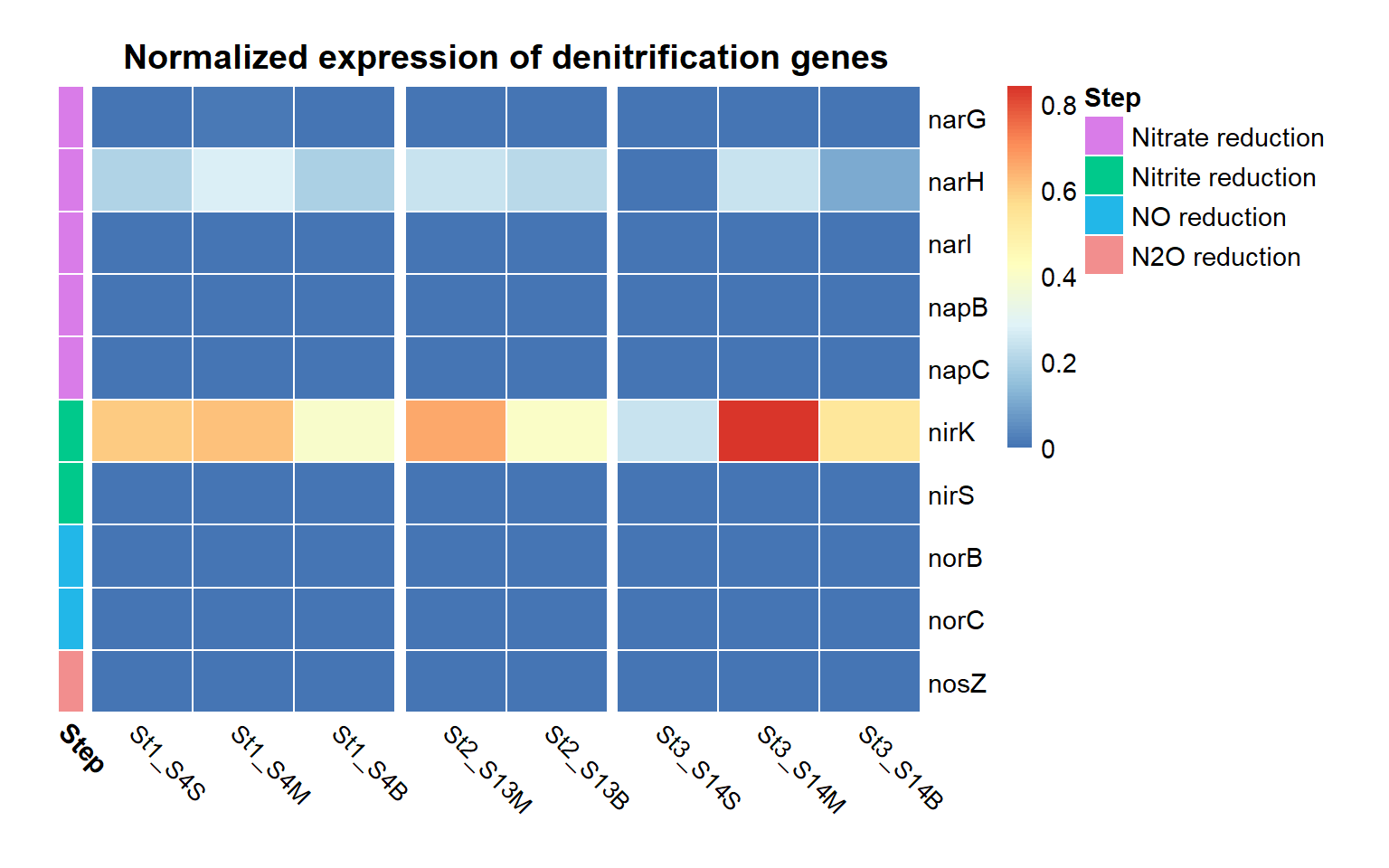
The heatmap shows the distribution and relative expression of denitrification-related KEGG orthologs (KO) genes across the stations and depth layers. Overall, the results indicate substantial spatial variability in the transcription of denitrification genes across the study area.
Nitrate reduction genes
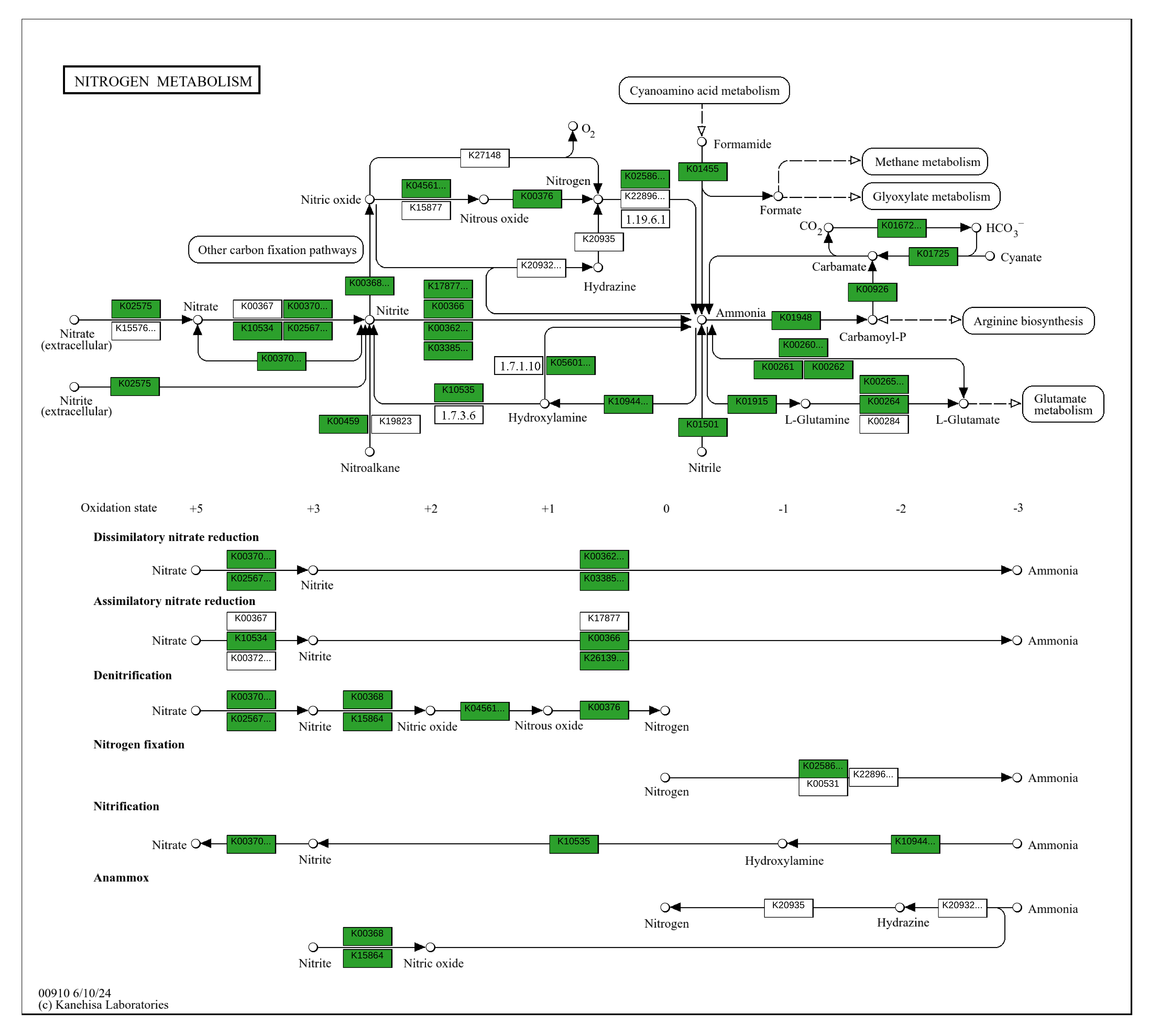
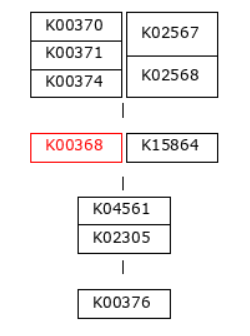
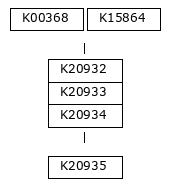
The first step of denitrification, the reduction of nitrate to nitrite, can be catalyzed by two distinct enzymatic systems: the membrane-bound nitrate reductase (Nar) and the periplasmic nitrate reductase (Nap). In this dataset, the Nar pathway is represented by K00370, K00371, and K00374, while the Nap pathway is represented by K02567 and K02568 (see pathway diagram above).
At Station 1, transcripts from both nitrate reduction systems (Nar and Nap) are detected.At Station 2, K00374 is absent, indicating only partial expression of the Nar-associated genes detected in this dataset, while with both the Nap genes K02567 and K02568, the Nap pathwa is complete. In contrast, Station 3 shows limited expression of Nar genes and Nap-associated transcripts are absent, indicating reduced transcription of nitrate-to-nitrite reduction pathways at this station.
Nitrite reduction genes
Genes associated with the reduction of nitrite to nitric oxide are represented by K00368 (nirK) and K15864 (nirS), which encode alternative nitrite reductases (the presence of only one transcript is enough). The gene of nirK exhibits the highest coverage across samples, indicating that the conversion of nitrite to nitric oxide may represent a dominant step in the denitrification pathway in these samples. In contrast, nirS shows considerably lower expression, suggesting a predominance of nirK-type denitrifiers in the active microbial assemblages.
Nitric oxide reduction genes
Genes involved in the reduction of nitric oxide to nitrous oxide are represented by K04561 (norB) and K02305 (norC), which are associated with the nitric oxide reductase system. The heatmap shows moderate transcript coverage at Stations 1 and 2, with the highest expression observed in the mid-water and bottom samples at Station 2. This suggests that the nitric oxide reduction step of denitrification is actively transcribed at Stations 1 and 2, particularly in deeper waters, whereas expression of nitric oxide reductase genes appears to be minimal at Station 3.
Nitrous oxide reduction gene
The final step of denitrification, the reduction of nitrous oxide to dinitrogen gas, is represented by K00376 (nosZ), alone. Transcripts of this gene are detected primarily at Stations 1 and 2, indicating that microorganisms capable of reducing nitrous oxide may be transcriptionally active in these environments. However, expression levels remain relatively moderate compared with earlier steps of the pathway, and transcripts are largely absent at Station 3, suggesting limited expression of this step at that station.
Depth-related patterns
Across Stations 1 and 2, several denitrification genes exhibit higher transcript levels in mid-water and bottom samples compared to surface waters, suggesting that denitrification activity may be enhanced in deeper parts of the water column.
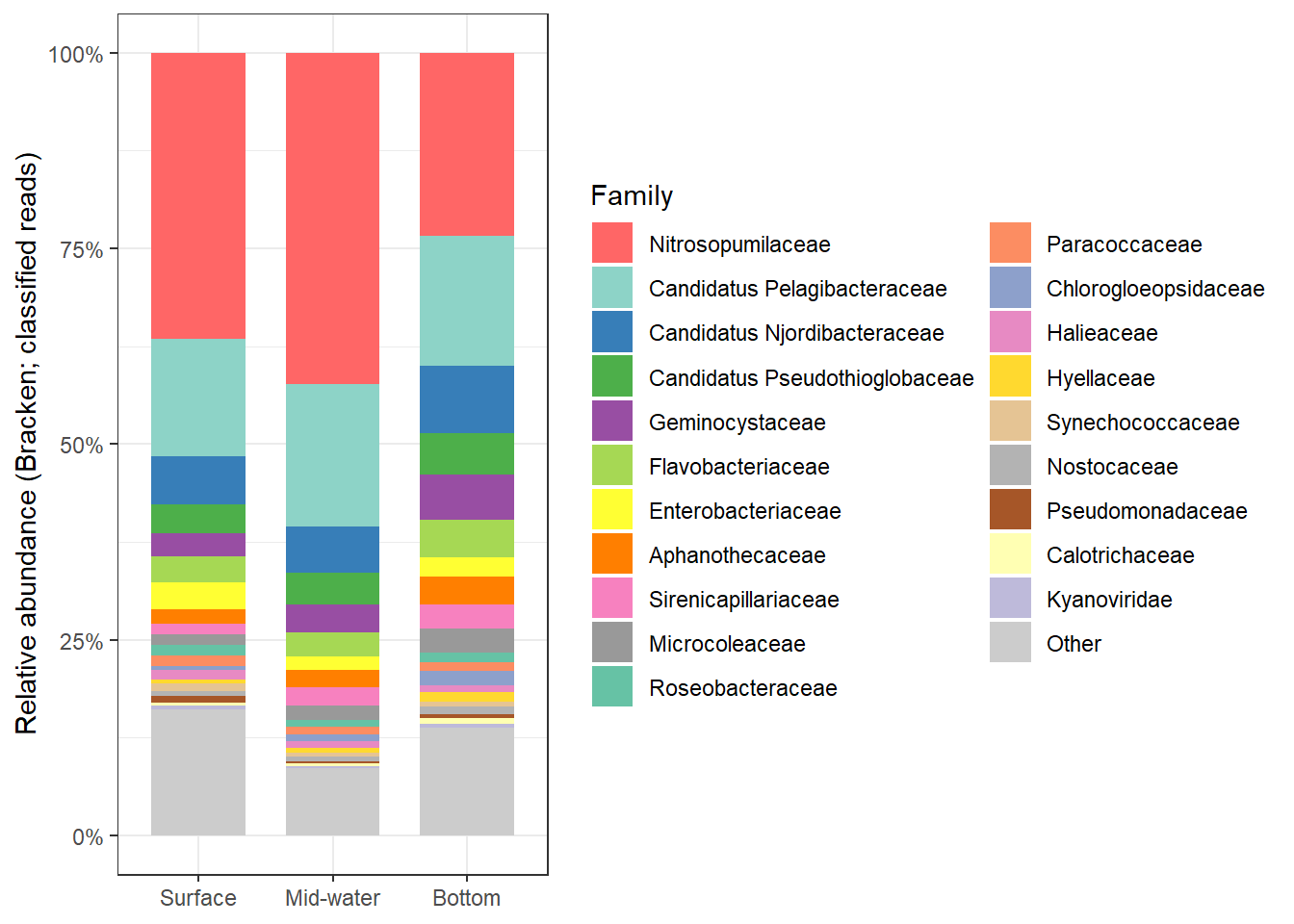
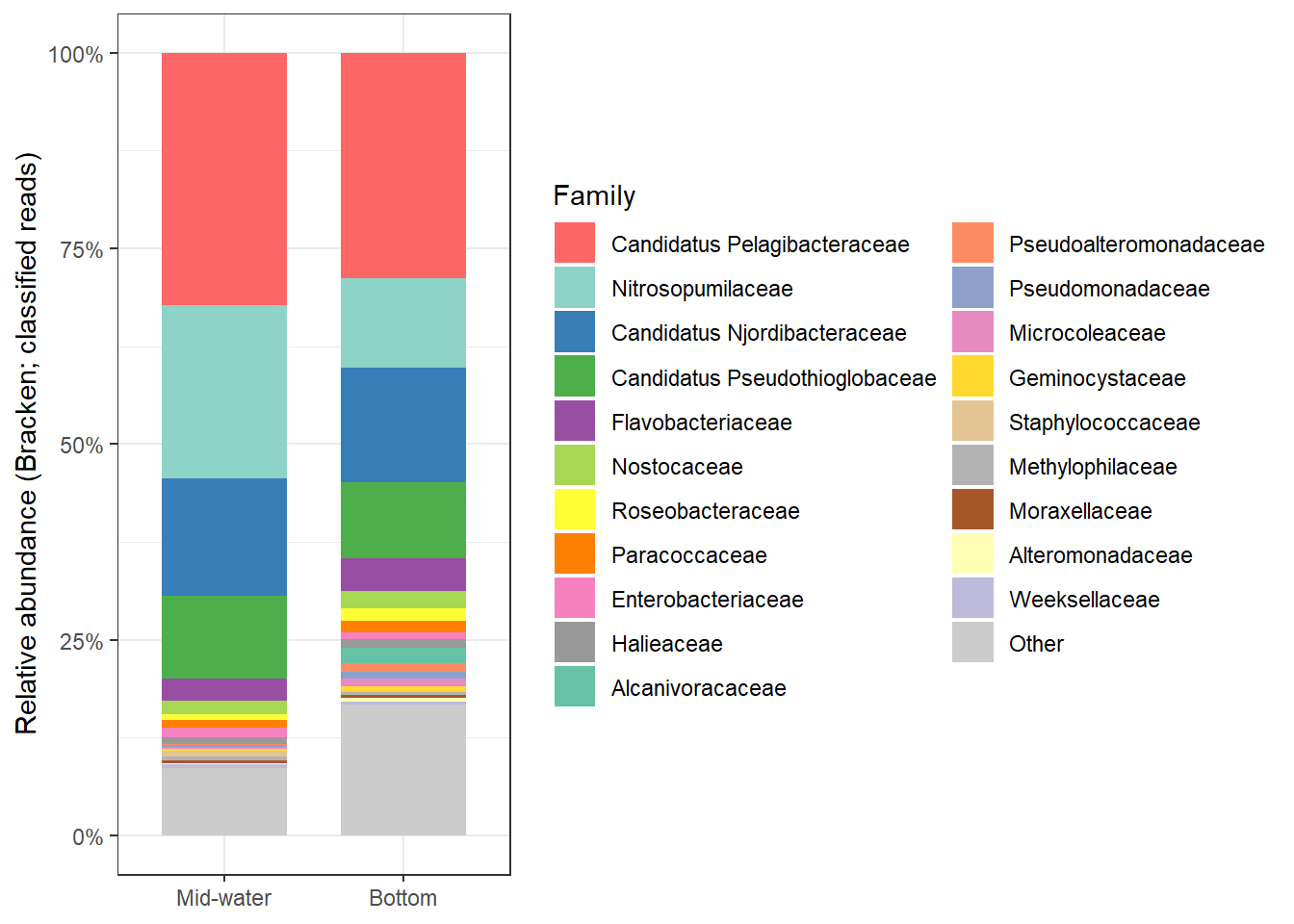
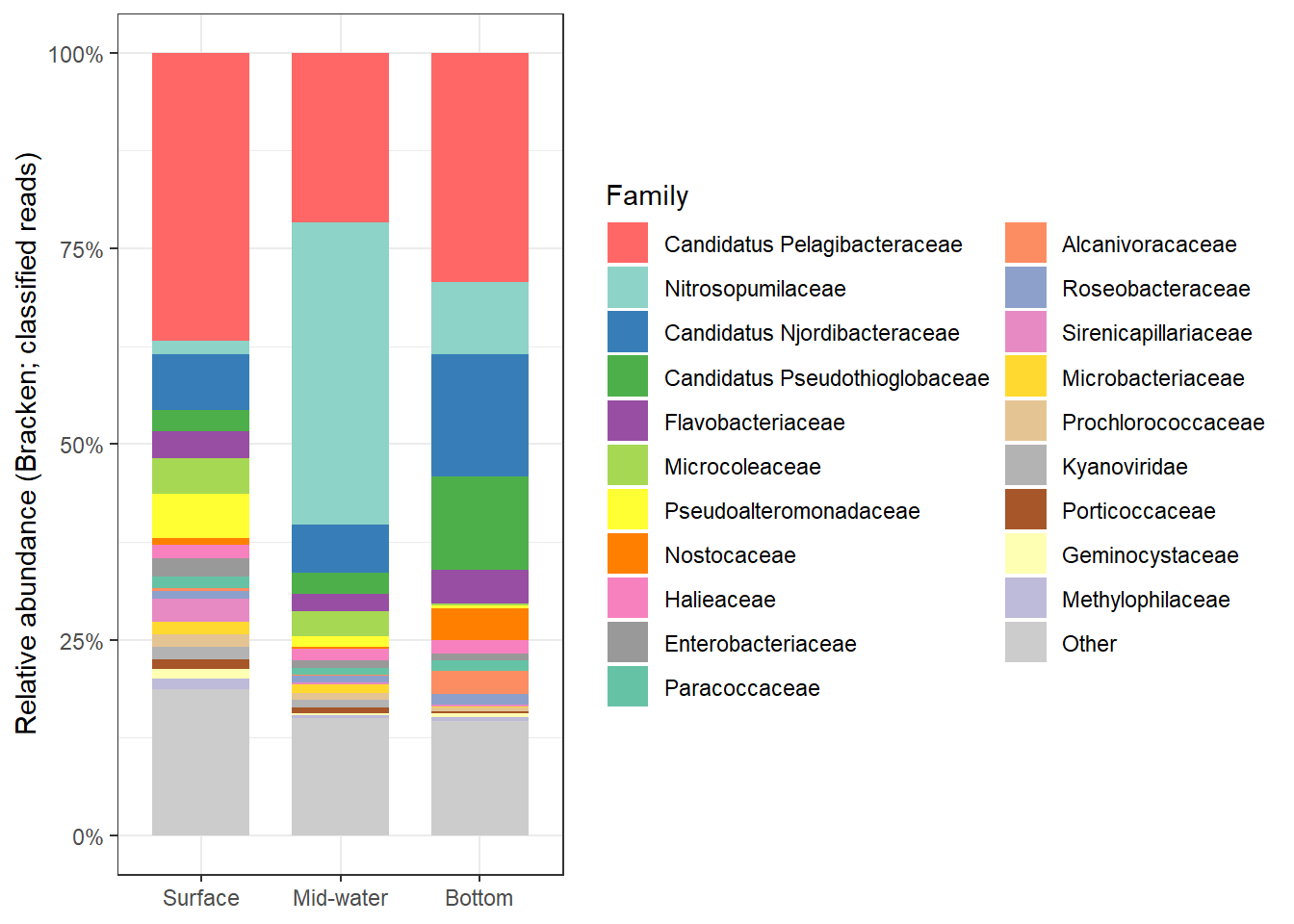
 2:45 AM on September 8, 2013 (UTC)
2:45 AM on September 8, 2013 (UTC)